REPAC: analysis of alternative polyadenylation from RNA-sequencing data

REPAC: analysis of alternative polyadenylation from RNA-sequencing data
Alternative polyadenylation (APA) is a crucial post-transcriptional regulatory mechanism that plays a significant role in gene expression, cellular differentiation, and the development of various diseases, including cancer and neurological disorders. Although there are specialized sequencing techniques available to analyze polyadenylation sites, the availability of such data remains limited compared to the more commonly used RNA-sequencing data. This gap poses a challenge for large-scale studies of APA, particularly in clinical and research settings.
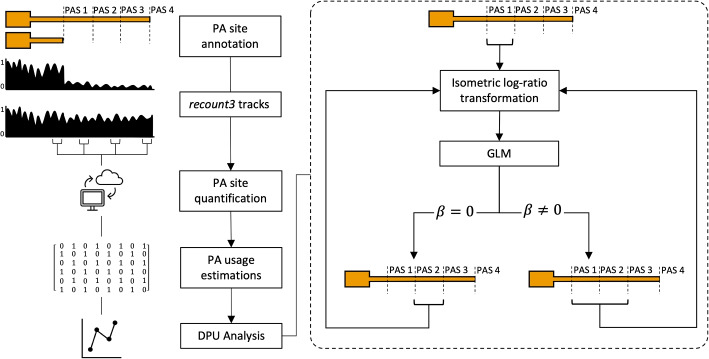
To overcome this limitation, we developed REPAC, an innovative framework that enables the analysis of APA directly from RNA-sequencing data. REPAC efficiently identifies and quantifies polyadenylation events without the need for specialized polyadenylation-specific sequencing methods. In this study, we apply REPAC to investigate the landscape of APA induced by the activation of B cells, providing insights into how APA contributes to cellular processes in immune response.
Additionally, we demonstrate that REPAC significantly outperforms other existing methods for APA analysis, showing a speed advantage of at least seven times faster, which makes it highly suitable for large-scale studies involving hundreds or even thousands of samples. The framework is designed to be computationally efficient and scalable, making it accessible for both small and large datasets. Furthermore, REPAC provides highly accurate results, offering an intuitive, user-friendly approach to APA analysis.
Overall, REPAC presents a powerful, accurate, and convenient solution for exploring the complexities of APA, enabling researchers to study its impact on gene regulation and disease with greater efficiency and precision.